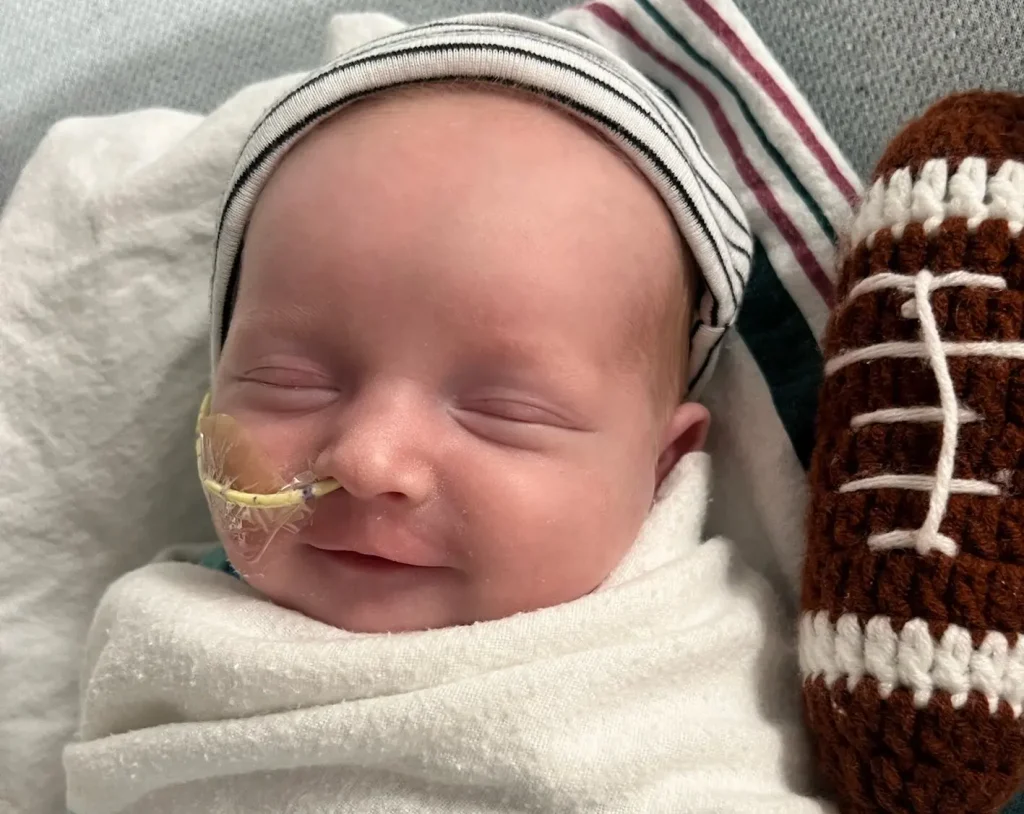
Λένε πως οι πιο δύσκολες μάχες δίνονται τις ήσυχες ώρες, και για τους Kyle και Nicole Muldoon, αυτές οι ώρες ξεκίνησαν τη στιγμή που το νεογέννητό τους έκλαψε για πρώτη φορά. Αντιμέτωποι με μια διάγνωση τόσο σπάνια και καταστροφική και με ελάχιστες διαθέσιμες θεραπείες υπήρχαν, ομάδα επιστημόνων και κλινικών ιατρών έσπευσε να αναπτύξει μια εξατομικευμένη θεραπεία με γονιδιακή τροποποίηση.
Αυτοάνοσα νοσήματα: Πώς σχετίζονται με τα γονίδια – Τι ρόλο παίζουν το φύλο και η κληρονομικότητα
Νέα εποχή στην ιατρική – Ομάδα επιστημόνων σώζει βρέφος με εξατομικευμένη θεραπεία με γονιδιακή τροποποίηση
Όταν το νεογέννητο των Kyle και Nicole Muldoon αντιμετώπισε ένα σχεδόν βέβαιο πεπρωμένο, οι γιατροί στο Παιδιατρικό Νοσοκομείο της Φιλαδέλφειας έδωσαν μια ζοφερή πρόγνωση: ο γιος τους, KJ, γεννήθηκε με ένα γενετικό ελάττωμα τόσο σπάνιο και σοβαρό που πολλοί γονείς επέλεγαν την άνεση αντί για την επιθετική θεραπεία. Όμως αυτή τη φορά, οι κλινικοί ιατροί και οι ερευνητές αρνήθηκαν να αποδεχτούν το κατεστημένο και σχεδίασαν εξατομικευμένη γονιδιακή θεραπεία.
Σε έναν αγώνα ενάντια στον χρόνο, κινητοποίησαν μια συμμαχία ακαδημαϊκών εργαστηρίων, βιοτεχνολογικών εταιρειών και ομοσπονδιακών ρυθμιστικών αρχών για να σχεδιάσουν μια μοναδική λύση γονιδιακής τροποποίησης προσαρμοσμένη στη συγκεκριμένη μετάλλαξη του KJ. Μέσα σε λίγους μήνες, το βρέφος θα λάμβανε μια πρωτοποριακή θεραπεία που θα αναδιαμόρφωνε το μέλλον της εξατομικευμένης ιατρικής.
Πώς χρησιμοποιείται το ανθρώπινο γονιδίωμα στην ιατρική, σύμφωνα με την έρευνα
Στη σημερινή ιατρική, τα δεδομένα του ανθρώπινου γονιδιώματος υποστηρίζουν μεταμορφωτικές προόδους σε πολλούς τομείς:
- Ογκολογία ακριβείας: Η εκτενής γονιδιωματική ανάλυση μέσω πάνελ αλληλούχισης νέας γενιάς είναι πλέον ρουτίνα, εντοπίζοντας χρήσιμες μεταλλάξεις σε πάνω από 80% των προχωρημένων καρκίνων και καθοδηγώντας τις συστάσεις των μοριακών συμβουλίων όγκου για σχεδόν 70% των ασθενών.
- Φαρμακογονιδιωματική: Τεστ ενός γονιδίου ή πάνελ — τυποποιημένα από CPIC και PharmGKB — χρησιμοποιούνται προληπτικά για την εξατομίκευση επιλογής φαρμάκων και δοσολογίας (π.χ. CYP2C19, HLA-B) και έχουν αποδεδειγμένα μειώσει τις ανεπιθύμητες αντιδράσεις ενώ βελτιώνουν την αποτελεσματικότητα.
- Γονιδιακή τροποποίηση: Η πρώτη εξατομικευμένη in-human θεραπεία βασικής τροποποίησης CRISPR για έλλειψη CPS1 διόρθωσε παθογόνο μετάλλαξη σε βρέφος έξι μηνών μέσω νανοσωματιδίων λιπιδίων, με αποτέλεσμα βελτίωση της μεταβολικής λειτουργίας και απόδειξη της εφικτότητας εξατομικευμένων θεραπειών τύπου N-of-1.
- Πολυγονιδιακοί δείκτες κινδύνου: Βελτιστοποιημένοι και επικυρωμένοι σε διάφορες καταγωγές, οι PRSs ταξινομούν τον ισόβιο κίνδυνο για παθήσεις όπως μεταβολική νόσος του ήπατος και καρδιαγγειακή νόσο, με κλινικά κανάλια να παράγουν πλέον εξατομικευμένες αναφορές κινδύνου για την καθοδήγηση πρώιμου ελέγχου και πρόληψης.
- Διάγνωση σπάνιων νόσων: Η αλληλούχιση ολόκληρου γονιδιώματος ως πρώτη γραμμή έχει αποφέρει διαγνωστικά ποσοστά ~29 %, αποκαλύπτοντας μεταλλάξεις σε κωδικοποιητικές, μη κωδικοποιητικές και δομικές περιοχές που δεν εντοπίζονται με την αλληλούχιση εξώματος, προκαλώντας αλλαγές στη διαχείριση σε έως και ένα τρίτο των περιπτώσεων.
- Μη επεμβατικός προγεννητικός έλεγχος: Με επέκταση πέρα από τις κοινές τρισωμίες σε σπάνιες αυτοσωμικές ανευπλοειδίες και μεταβολές αριθμού αντιγράφων, βαθύτερες προσεγγίσεις αλληλούχισης έχουν βελτιώσει σημαντικά τις θετικές προβλεπτικές τιμές για παθήσεις όπως η τρισωμία 18, ενισχύοντας την προγεννητική αξιολόγηση κινδύνου και συμβουλευτική.
Μαζί, αυτές οι στρατηγικές που καθοδηγούνται από το γονιδίωμα εισάγουν μια νέα εποχή εξατομικευμένης, προβλεπτικής και προληπτικής υγειονομικής περίθαλψης.
Βρέφος με σπάνια έλλειψη CPS1 θεραπεύεται με νέα εξατομικευμένη θεραπεία γονιδιακής τροποποίησης
Κάτι ήταν πολύ λάθος με το μωρό των Kyle και Nicole Muldoon. Οι γιατροί υπέθεσαν. Ίσως μηνιγγίτιδα; Ίσως σήψη; Πήραν απάντηση όταν ο KJ ήταν μόλις μιας εβδομάδας. Είχε μια σπάνια γενετική διαταραχή, την έλλειψη CPS1, που επηρεάζει μόλις ένα στα 1,3 εκατομμύρια μωρά. Αν επιζούσε, θα είχε σοβαρές νοητικές και αναπτυξιακές καθυστερήσεις και τελικά θα χρειαζόταν μεταμόσχευση ήπατος. Όμως τα μισά βρέφη με αυτή τη διαταραχή πεθαίνουν την πρώτη εβδομάδα ζωής.

Οι γιατροί στο Παιδιατρικό Νοσοκομείο της Φιλαδέλφειας προσέφεραν στους Muldoon φροντίδα με σκοπό την άνεση και όχι τη θεραπεία για το μωρό τους, την ευκαιρία να αποφύγουν τις επιθετικές θεραπείες μπροστά σε μια ζοφερή πρόγνωση. «Τον αγαπούσαμε και δεν θέλαμε να υποφέρει», είπε η κα Muldoon, σύμφωνα με το New York Times. Όμως εκείνη και ο σύζυγός της αποφάσισαν να δώσουν στον KJ μια ευκαιρία.
Αντίθετα, ο KJ έγραψε ιστορία στην ιατρική. Το μωρό, πλέον 9 ½ μηνών, έγινε ο πρώτος ασθενής κάθε ηλικίας που έλαβε εξατομικευμένη θεραπεία γονιδιακής τροποποίησης, σύμφωνα με τους γιατρούς του. Έλαβε μια έγχυση φτιαγμένη αποκλειστικά γι’ αυτόν και σχεδιασμένη να διορθώσει τη συγκεκριμένη του μετάλλαξη.
Νέο μοντέλο για τη θεραπεία σπάνιων γενετικών νοσημάτων
Οι ερευνητές που ηγήθηκαν της προσπάθειας να σωθεί ο KJ παρουσιάζουν την εργασία τους την Πέμπτη στο ετήσιο συνέδριο της American Society of Gene & Cell Therapy και τη δημοσιεύουν επίσης στο New England Journal of Medicine. Οι επιπτώσεις της θεραπείας ξεπερνούν κατά πολύ τη θεραπεία του KJ, δήλωσε ο Αμερικανός αιματολόγος Dr Peter Marks, ο οποίος ήταν ο αρμόδιος στον Οργανισμό Τροφίμων και Φαρμάκων για τη ρύθμιση της γονιδιακής θεραπείας μέχρι που παραιτήθηκε πρόσφατα λόγω διαφωνιών με τον Robert F. Kennedy Jr., τον υπουργό υγείας και ανθρώπινων υπηρεσιών.
Πάνω από 30 εκατομμύρια άνθρωποι στις Ηνωμένες Πολιτείες έχουν μία από τις περισσότερες από 7.000 σπάνιες γενετικές ασθένειες. Στην Ελλάδα, οι σπάνιες ασθένειες πλήττουν συνολικά περίπου 350.000–600.000 άτομα, ενώ στην Ευρώπη ορίζονται ως παθήσεις που αφορούν λιγότερα από 5 άτομα ανά 10.000 και συλλογικά επηρεάζουν το 6–8% του πληθυσμού. Οι περισσότερες είναι τόσο σπάνιες που καμία εταιρεία δεν είναι πρόθυμη να δαπανήσει χρόνια για την ανάπτυξη μιας γονιδιακής θεραπείας που θα χρειάζονταν τόσο λίγοι άνθρωποι.
Όμως η θεραπεία του KJ — που βασίστηκε σε δεκαετίες έρευνας με χρηματοδότηση από το κράτος των ΗΠΑ — προσφέρει ένα νέο μονοπάτι για τις εταιρείες να αναπτύσσουν εξατομικευμένες θεραπείες χωρίς να περάσουν από χρόνια δαπανηρής ανάπτυξης και δοκιμών.
Ασθένειες όπως του KJ είναι αποτέλεσμα μιας μόνο μετάλλαξης — ενός λανθασμένου γράμματος DNA ανάμεσα στα τρία δισεκατομμύρια του ανθρώπινου γονιδιώματος. Η διόρθωσή του απαιτεί ακριβή στόχευση σε μια προσέγγιση που ονομάζεται βασική τροποποίηση.
Για να επιτευχθεί αυτό το κατόρθωμα, η θεραπεία ενθυλακώνεται σε λιπαρά μόρια λιπιδίων ώστε να προστατευθεί από αποδόμηση στο αίμα καθ’ οδόν προς το ήπαρ, όπου θα γίνει η διόρθωση. Μέσα στα λιπίδια βρίσκονται οδηγίες που δίνουν εντολή στα κύτταρα να παράγουν ένα ένζυμο που τροποποιεί το γονίδιο. Μεταφέρουν επίσης ένα μοριακό GPS — το CRISPR — το οποίο τροποποιήθηκε ώστε να «σέρνεται» πάνω στο DNA ενός ανθρώπου μέχρι να βρει το ακριβές γράμμα DNA που πρέπει να αλλάξει.
Η θεραπεία του KJ μπορεί να γίνει βάση για εξατομικευμένες θεραπείες και άλλων ασθενών με σπάνιες νόσους
Ενώ η θεραπεία του KJ προσαρμόστηκε ώστε το CRISPR να εντοπίζει μόνο τη δική του μετάλλαξη, η ίδια μέθοδος θα μπορούσε να προσαρμοστεί και να χρησιμοποιηθεί ξανά και ξανά για να διορθώσει μεταλλάξεις σε άλλα σημεία του DNA ενός ατόμου. Μόνο οι οδηγίες του CRISPR που οδηγούν τον τροποποιητή στο σημείο της μετάλλαξης στο DNA θα χρειάζονταν αλλαγή. Οι θεραπείες θα ήταν φθηνότερες, «τουλάχιστον κατά μία τάξη μεγέθους», δήλωσε ο Dr Marks.
Γονίδια και καρκίνος: Τι συμβαίνει πριν από τη γέννηση και αυξάνει τον κίνδυνο
Η μέθοδος, δήλωσε ο Dr Marks, ο οποίος έγραψε ένα editorial συνοδευτικό της ερευνητικής δημοσίευσης, «είναι, για εμένα, μία από τις πιο δυνητικά μεταμορφωτικές τεχνολογίες που υπάρχουν». Τελικά, θα μπορούσε επίσης να χρησιμοποιηθεί για πιο κοινές γενετικές διαταραχές όπως η δρεπανοκυτταρική νόσος, η κυστική ίνωση, η νόσος του Huntington και η μυϊκή δυστροφία. Και, είπε, θα μπορούσε «πραγματικά να μεταμορφώσει την υγειονομική περίθαλψη».

Η ιστορία της εξατομικευμένης θεραπείας γονιδιακής τροποποίησης του KJ ξεκίνησε το βράδυ της 8ης Αυγούστου, όταν ο Dr Kiran Musunuru, ερευνητής γονιδιακής τροποποίησης στο University of Pennsylvania, έλαβε ένα email από την Dr Rebecca Ahrens-Nicklas στο Children’s Hospital of Philadelphia. Είχε γεννηθεί ένα μωρό και οι γενετικές εξετάσεις έδειξαν ότι είχε έλλειψη CPS1. Μπορούσε να σώσει το μωρό;
Ταχεία η ανάπτυξη προσαρμοσμένου τροποποιητή γονιδίων για τον KJ
Ο Dr Musunuru είχε αρχίσει να ερευνά τη χρήση της γονιδιακής τροποποίησης για σχετικά κοινές μεταλλάξεις. Η ανάπτυξη ενός τροποποιητή γονιδίων για θεραπεία ασθενών είναι μια σκόπιμη διαδικασία που μπορεί να διαρκέσει χρόνια. Όμως ο KJ δεν είχε χρόνια να περιμένει — ίσως μόνο έξι μήνες πριν από έναν αυξανόμενο κίνδυνο σοβαρής εγκεφαλικής βλάβης ή θανάτου.
«Σε αυτό το σημείο, ξεκινά το ρολόι στο μυαλό μου», είπε ο Dr Musunuru. «Αυτή είναι η πραγματική ζωή. Δεν είναι θεωρητικό».
Η ασθένεια του KJ προκαλείται από ανικανότητα του οργανισμού να αποβάλει την αμμωνία, ένα υποπροϊόν του μεταβολισμού πρωτεΐνης. Η αμμωνία συσσωρεύεται στο αίμα και περνά στον εγκέφαλο. Οι γιατροί του τον έβαλαν σε δίαιτα που περιόριζε αυστηρά την πρωτεΐνη — μόλις αρκετή για να αναπτυχθεί. Είχε επίσης ένα φάρμακο, τη φαινυλοβουτυρική γλυκερόλη, που βοηθούσε στην απομάκρυνση της αμμωνίας από το αίμα του. Όμως παρέμενε σε υψηλό κίνδυνο για εγκεφαλική βλάβη ή θάνατο.
Οποιαδήποτε ασθένεια ή λοίμωξη θα μπορούσε να εκτοξεύσει τα επίπεδα αμμωνίας και να προκαλέσει μη αναστρέψιμη βλάβη στον εγκέφαλο. Ο KJ ζούσε στο νοσοκομείο υπό 24ωρη φροντίδα. Η δημιουργία και η δοκιμή ενός συστήματος γονιδιακής τροποποίησης για το μωρό των Muldoon δεν ήταν εύκολη. «Υπήρχε πολλή δουλειά χωρίς συγκεκριμένο σχέδιο», είπε ο Dr Musunuru.
Συμμαχία ακαδημαϊκών και βιομηχανίας επιταχύνει την ανάπτυξη CRISPR κλινικής ποιότητας
Ξεκίνησε να συνεργάζεται με τον Ρώσο Fyodor Urnov στο University of California, Berkeley, ο οποίος φρόντισε να μην υπάρχουν απρόσμενες και επιζήμιες τροποποιήσεις γονιδίων σε άλλα σημεία του DNA. Ο Dr Urnov είναι μέρος μιας ακαδημαϊκής συνεργασίας με την εταιρεία Danaher Corporation, μια εταιρεία ικανή να παράγει τον τροποποιητή γονιδίων για τον KJ με πρότυπα που θα επέτρεπαν τη χρήση του σε ασθενή.

Η Danaher με τη σειρά της συνεργάστηκε με δύο άλλες θυγατρικές της εταιρείες, δύο επιπλέον βιοτεχνολογικές εταιρείες και ένα ακόμη ερευνητικό ινστιτούτο, δήλωσε ο Αμερικανός Sadik Kassim, επικεφαλής τεχνολογίας για γονιδιακά φάρμακα της εταιρείας.
«Σε κάθε βήμα της διαδικασίας, πάντα περιμέναμε κάποιος να πει “Όχι, συγγνώμη”», δήλωσε ο Dr Kassim. «Και τότε θα ήταν το τέλος της ιστορίας». Όμως οι φόβοι του δεν επιβεβαιώθηκαν. Η Danaher και οι άλλες εταιρείες χρέωσαν μόνο τα πρώτες ύλες για την παραγωγή του φαρμάκου, πρόσθεσε. Ο FDA επίσης διευκόλυνε την κανονιστική έγκριση της θεραπείας, είπε η Dr Ahrens-Nicklas, παιδίατρος στη Philadelphia.
Δεκάδες ερευνητές άφησαν όλα τα άλλα στην άκρη για μήνες. Στο Berkeley, ο Dr Urnov είπε, «οι επιστήμονες έκαιγαν ένα βαρέλι νυχτερινό λάδι στο μέγεθος του κόλπου του Σαν Φρανσίσκο». Πρόσθεσε ότι «τέτοια ταχύτητα στην παραγωγή CRISPR κλινικής ποιότητας για γενετική ασθένεια δεν έχει προηγούμενο στον τομέα μας. Δεν ήταν καν κοντά».
Ο David Liu από το Harvard, του οποίου το εργαστήριο εφηύρε τη μέθοδο γονιδιακής τροποποίησης που χρησιμοποιήθηκε για τη διόρθωση της μετάλλαξης του KJ, είπε ότι η ταχύτητα ήταν «εκπληκτική». «Αυτά τα βήματα συνήθως διαρκούν το μεγαλύτερο μέρος μιας δεκαετίας, αν όχι περισσότερο», είπε.
Γονική συναίνεση, κλιμάκωση δόσης και πρώιμα κλινικά ορόσημα για τη θεραπεία του βρέφους
Μόνο όταν είχαν τη λύση γονιδιακής τροποποίησης στα χέρια τους και ο FDA ενέκρινε την εργασία των ερευνητών, η Dr Ahrens-Nicklas πλησίασε τους γονείς του KJ. «Μία από τις πιο τρομακτικές στιγμές ήταν όταν μπήκα στο δωμάτιο και είπα, “Δεν ξέρω αν θα λειτουργήσει αλλά υπόσχομαι ότι θα κάνω τα πάντα για να είναι ασφαλές”», είπε.
Το πρωί της 25ης Φεβρουαρίου, ο KJ έλαβε την πρώτη έγχυση, μια πολύ χαμηλή δόση επειδή κανείς δεν ήξερε πώς θα αντιδρούσε. Ήταν στο δωμάτιό του, στην κούνια όπου είχε ζήσει όλη του τη ζωή. Ήταν 6 μηνών και βρισκόταν στο έβδομο εκατοστημόριο του βάρους του.
Ο Dr Musunuru παρακολούθησε την δίωρη έγχυση, νιώθοντας, όπως είπε, «και ενθουσιασμένος και τρομοκρατημένος». Ο KJ κοιμήθηκε καθ’ όλη τη διάρκεια της διαδικασίας. Μέσα σε δύο εβδομάδες, ο KJ μπορούσε να τρώει τόση πρωτεΐνη όσο ένα υγιές μωρό. Όμως συνέχισε να χρειάζεται το φάρμακο για την απομάκρυνση της αμμωνίας από το αίμα του — ένα σημάδι ότι ο τροποποιητής δεν είχε ακόμη διορθώσει το DNA σε κάθε προσβεβλημένο κύτταρο. Οι γιατροί του έδωσαν δεύτερη δόση 22 ημέρες αργότερα.
Κατάφεραν να μειώσουν στο μισό τη δόση του φαρμάκου. Έπαθε μερικές ιογενείς λοιμώξεις εκείνη την περίοδο, οι οποίες υπό κανονικές συνθήκες θα προκαλούσαν τρομακτικά ξεσπάσματα στα επίπεδα αμμωνίας. Όμως, όπως είπε η Dr Ahrens-Nicklas, «τις πέρασε άψογα». Μιάμιση εβδομάδα πριν, η ομάδα έδωσε στον KJ και τρίτη δόση.
Είναι πολύ νωρίς για να γνωρίζουν αν μπορεί να σταματήσει τελείως τη φαρμακευτική αγωγή, αλλά η δοσολογία έχει μειωθεί σημαντικά. Και είναι αρκετά καλά για να ξεκινήσει η ομάδα τον σχεδιασμό εξόδου του από το νοσοκομείο. Πιάνει τα αναπτυξιακά του ορόσημα και το βάρος του βρίσκεται πλέον στο 40ό εκατοστημόριο για την ηλικία του, αλλά δεν είναι ακόμα γνωστό αν θα αποφύγει τη μεταμόσχευση ήπατος.
Από τη θεμελιώδη έρευνα CRISPR σε σωτήρια θεραπεία
Το αποτέλεσμα «είναι ένας θρίαμβος για την επένδυση του αμερικανικού λαού στην βιοϊατρική έρευνα», δήλωσε ο Dr Urnov. Οι ερευνητές τόνισαν τον ρόλο της κρατικής χρηματοδότησης στην ανάπτυξη. Η εργασία, όπως είπαν, ξεκίνησε δεκαετίες πριν με ομοσπονδιακή χρηματοδότηση για βασική έρευνα στα βακτηριακά ανοσοποιητικά συστήματα. Αυτό οδήγησε τελικά, με επιπλέον κρατική υποστήριξη, στην ανακάλυψη του CRISPR. Η ομοσπονδιακή επένδυση στην αλληλούχιση του ανθρώπινου γονιδιώματος κατέστησε δυνατή την ταυτοποίηση της μετάλλαξης του KJ.
4 αλλαγές που μπορούν να νικήσουν τα «κακά» γονίδια και να χαρίσουν 5 χρόνια ζωής
Η κρατική χρηματοδότηση υποστήριξε το εργαστήριο του Dr Liu και την ανακάλυψη της μεθόδου τροποποίησης. Ένα ομοσπονδιακό πρόγραμμα μελέτης της γονιδιακής τροποποίησης στήριξε την έρευνα του Dr Musunuru. Παράλληλα, υπήρχε κρατικά χρηματοδοτούμενη εργασία που οδήγησε στην κατανόηση της νόσου του KJ. «Δεν νομίζω ότι αυτό θα μπορούσε να συμβεί σε καμία άλλη χώρα εκτός από τις ΗΠΑ», είπε ο Dr Urnov. Όσοι εργάστηκαν για να σώσουν τον KJ ένιωθαν υπερηφάνεια, είπε ο Dr Urnov. «Όλοι είπαμε ο ένας στον άλλο, “Αυτό είναι το πιο σημαντικό πράγμα που έχουμε κάνει ποτέ”».
Πηγές: NYTimes, Nature, PubMed, AnnualReviews, Penn Medicine, Nature, NPJ, Scientific Reports, NEJM